
P3: Multiscale simulations of single site catalysis in porous materials
Single site catalysts (SAC) are currently intensely investigated as alternatives to homogeneous catalysts, because of their high specific activity and selectivity. In order to function properly, SAC requires a well-controlled environment. In this project, we focus on metal-organic frameworks (MOFs) and other porous materials as a model systems for SAC. MOFs have a well-defined structure and easily tunable porosity. They are hybrid materials comprising metal (or metal-oxo) nodes, known as secondary building units (SBUs), and organic linkers. Due to the diversity of SBUs and linkers, MOF allows the design of catalytic active sites with a precision approaching that of molecular catalysts while keeping high structure stability, easy product separation, and reversibility.
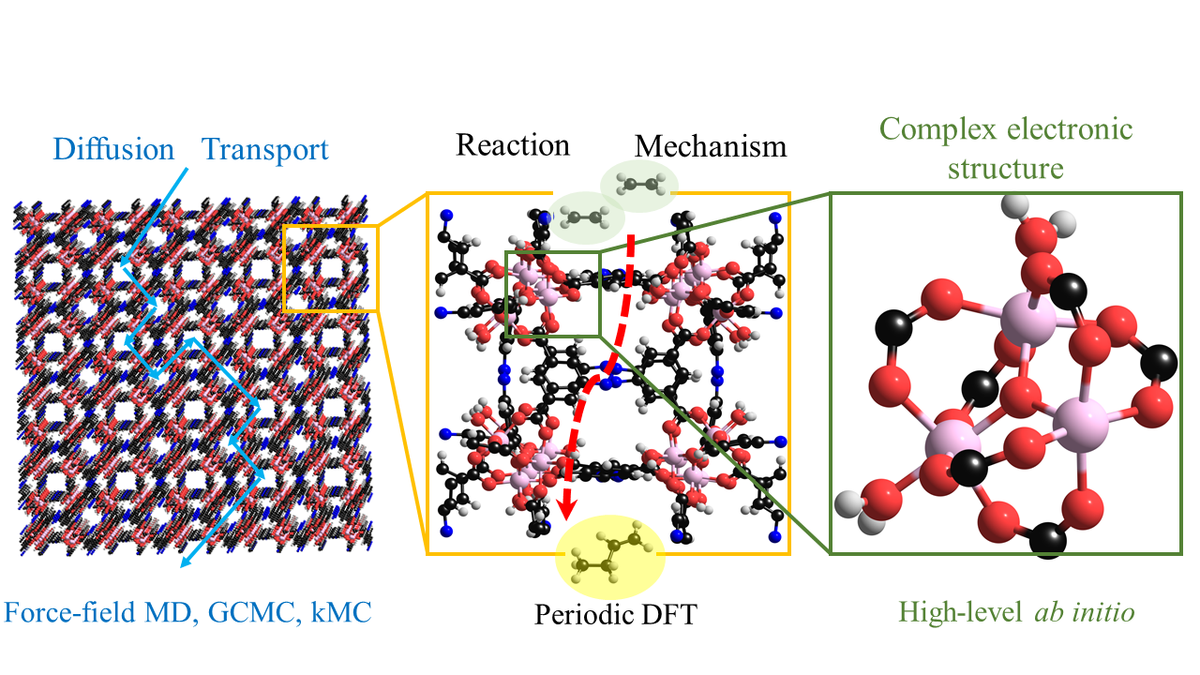
Our aim is develop and apply scale-bridging methods to calculate catalytic activity and selectivity of MOFs and other porous materials considering nanostructure confinement effects, diffusion and high-level quantum mechanical (QM) effects. We chose the oligomerization of ethylene to longer chain olefins as a test reaction as it is highly relevant for processes of the renewable chemical industry.
For improvement of SAC materials, we investigate: i) the influence of pore confinement, ii) the diffusion, i.e. mass transfer, of reactants, and iii) the selectivity of reactions.
Thus, we work on the development of a hierarchical protocol comprising methods from three different levels of theory. High-level quantum mechanics and density functional theory (DFT) are used to calculate reaction rates, while all-atom molecular dynamics (AA-MD) and Monte Carlo (MC) simulations are utilized to calculate adsorption and diffusion of reactants. Using embedded cluster approaches, we characterize the electronic structure of catalytic centers, benchmark methods to study reactions in MOF pores using DFT and recalculate reaction energies and barriers by higher level methods. These investigations are complemented with higher-scale models for adsorption and transport of educts and products through the porous materials, which ultimately allow consideration the macrokinetics of the process.
| Name | Title | Group | Contact |
|---|---|---|---|
| Avdoshin, Aleksandr | Prof. Wolfgang Wenzel / Dr. Mariana Kozlowska, KIT INT (P3) | aleksandr avdoshin ∂does-not-exist.kit edu 640 |
|
| Matsokin, Nikita | PD Dr. Karin Fink, KIT INT (P3) | +49 721 608-26974 nikita matsokin ∂does-not-exist.kit edu CN 640 202 |