
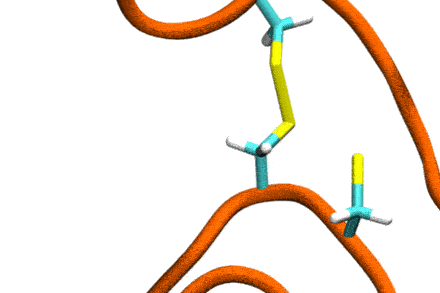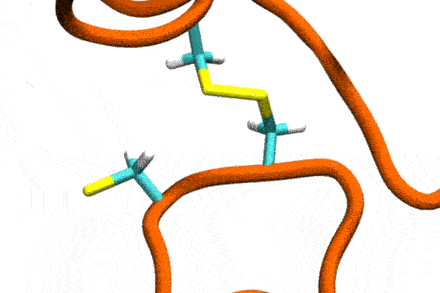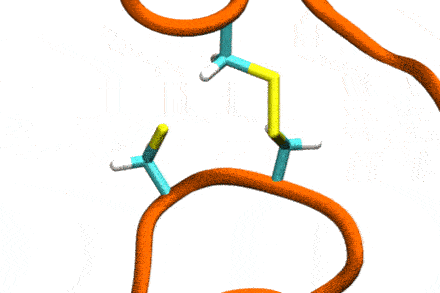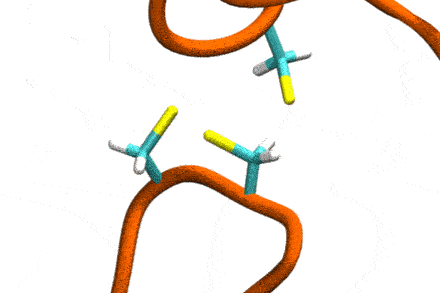
P4: Disulfide Shuffling in Proteins on Multiple Scales
Studies increasingly report on the labile nature of disulfide bonds in various enzymes, meaning that the different cysteine residues involved in disulfide bonding exchange partners dynamically. These processes may, for instance, take place in response to external mechanical stresses, or take part actively in the working cycle of the enzyme. An example is the labile disulfide bonds in the von Willebrand factor – a blood coagulation factor, which self-assembles (among others) through disulfide bond shuffling in its C-type domains. The essence of the process is a nucleophilic attack of a free cysteine thiolate onto an existing disulfide bond.
Since this reaction can only take place if the orientation of the reactants is convenient, the reaction is coupled to the conformational dynamics of the protein. Clearly, there is a large separation between these processes: The reaction is localized in a smart part of the protein and proceeds rather fast (if it proceeds at all). On the other hand, the relevant conformational changes may involve all of the large protein, and may take place on a largely extended temporal scale. Understandably, so far, no computational scheme has been developed which would allow to simulate these processes in an integrated fashion.
The aim of this project is to construct a recursive, multi-scale computational scheme to simulate – generally – chemical reactions coupled to conformational dynamics of, e.g., a protein. In a first step, the scheme will be developed and tested for disulfide shuffling in proteins. The method will take advantage of atomistic force fields for proteins as well as of Gō-type coarse-grained models, and will introduce chemical reactivity on the basis of QM/MM free energy calculations. This way, accuracy shall be maximized with computational complexity being minimized. The implementation will involve loosely coupled recursive schemes, will apply work-flow technology from project P6, and will implement accelerated MD integrators from project P7.
| Name | Title | Group | Contact |
|---|---|---|---|
| Vogelbacher, Maike | M. Sc. | Prof. Marcus Elstner, KIT IPC (associated) | maike vogelbacher ∂does-not-exist.kit edu CS 30.44 |