Projects
The common theme of all problems addressed in the RTG is concerned with the foundations of simulation methods from atomistic to mesoscopic resolution. Chemical details matter, and the question is, how those affect macroscopic properties and vice versa, how macroscopic properties steer processes at the molecular level. These problems exhibit an inherent recursiveness, coupling processes on different time-and/or length-scales.
Please click on the projects below for more detailed information:
P1: Adhesion and Friction between Solid and Soft Matter at Multiple Scales
P2: Degradation Mechanisms in Organic Electronics
P3: Multiscale simulations of single site catalysis in porous materials
P4: Disulfide Shuffling in Proteins on Multiple Scales
P5: A Multiscale Treatment of Kinase Function
P6: Scalable Computing Techniques
P7: Accelerated Sampling Techniques
P8: Graph Neural Network Based Multi-Agent Learning for Hierarchical Adaptive Coarse-Graining
Interdisciplinarity in the RTG
Collaborations
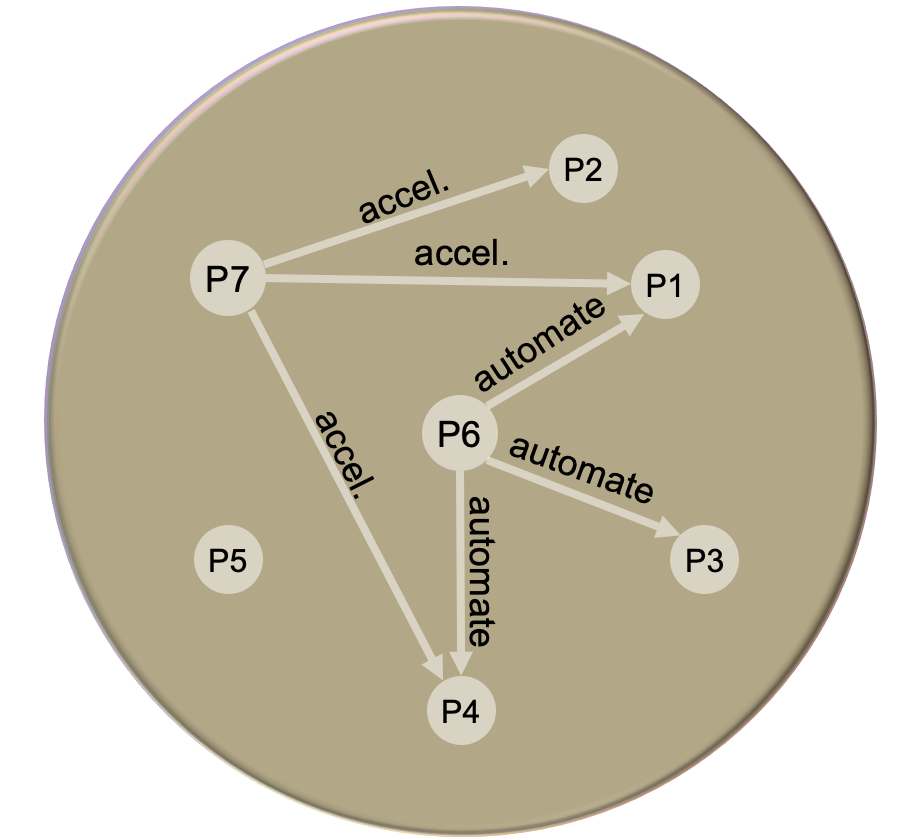
Addressing research problems that span multiple scales often requires a broad set of methodological competences and resources from various disciplines and teams. Cross-disciplinary collaboration is an important pillar of the RTG. For example, the implementation of work-flow techniques had an important role for the integration of methods from different scales. Furthermore, the development of efficient integrators (P7) helps accelerate the classical MD simulations which play a role in various projects of the RTG.
An example: The collaboration between P1, P6 and P7
The numerical stability of explicit time integrators, typically used in molecular dynamics simulations, is limited to a few femtoseconds. The upper limit of the time step size is even lowered when considering molecular rather than simple monatomic fluids. Therefore, the aim of the P1-P7 collaboration is to use of an explicit, time-reversible and parallelizable integrator that is stable at large time step sizes for large scale simulations. By using the Gautschi integrator scheme [1], provided and implemented in LAMMPS1 by P7, stable simulations with larger time step sizes were achieved. The efficiency and transferability of the integrator was demonstrated with the equation of state, thermal conductivity and surface tension of n-alkane fluids.
Another challenge is to find numerical solutions for the stress tensor of nano-confined fluids obtained through non equilibrium molecular dynamics (NEMD) simulations. The main aim is to understand the effects of the applied external loads, the contacting surfaces-lubricant fluid interaction, the method of driving the lubricant atoms (shear-driven, pressure-driven or both) and several other parameters on the lubricant’s viscosity, temperature and stress state. Therefore, a workflow management tool is crucial to explore the wide parametric space of nano-confined fluids and help with the coarse- grained molecular dynamics-continuum scale coupling. A FireWorks2 workflow was created and can be easily adapted to a config file for wfgenes3, which provides a lightweight framework that supports different input languages.
References
[1] Marlis Hochbruck and Alexander Ostermann. Exponential integrators. Acta Numerica, 19:209 – 286, 2010.