
P5: A Multiscale Treatment of Kinase Function
Kinase enzymes are a key component in various biochemical processes. Our aim in this project is to investigate the mechanism of such enzymatic reaction in a suitable computational method.
Enzymes functions are delicately balanced interplay of protein component motions and chemical reactions. Unfortunately, traditional computational methods struggle to describe these coupled motions of chemical reactions and at the same time protein side chain motions. While some studies focus on the quantum chemical description of the chemical reaction in a stationary (QM) or near-stationary (QM/MM) environment corresponding to a defined protein conformation, other studies focus on investigating the large-scale conformational transitions using molecular mechanics or coarse-grained force fields.
The main objective of P5 is to develop a transferable multiscale computational framework that is suitable for the simulation of enzymatic processes and the linked large-scale conformational transitions. Our model system is sensor histidine kinases (HK) which function through a reversible interplay of conformational transitions between two states, an autophosphorylation reaction and a phosphoryl-transfer reaction with a response regulator (RR) protein. Two-component systems (TCS), one of the main signal transduction mechanisms in bacteria, are comprised of HK and RR proteins. Once a stimulus is detected by the periplasmic sensor domain of HK, this signal is propagated along the multidomain, transmembrane HK via a series of conformational transitions ruled by unstable transient interactions. The dimerization histidine phosphotransfer (DHp) domain, contains a phosphorylatable conserved histidine residue. The catalytic ATP-binding (CA) domain binds ATP and hence provides the phosphoryl group necessary for phosphorylating the histidine residue in the DHp. The phosphoryl group from the histidine residue of the DHp is then transferred to an aspartate residue of the RR protein.
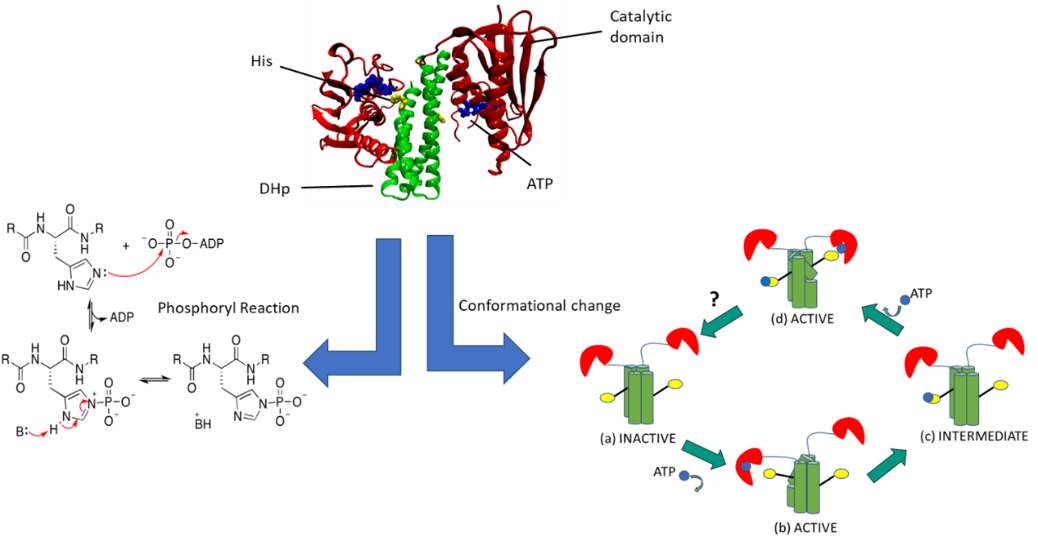
Our study can be classified into major two parts:
(A) Coarse-grained Method : Conformational Change
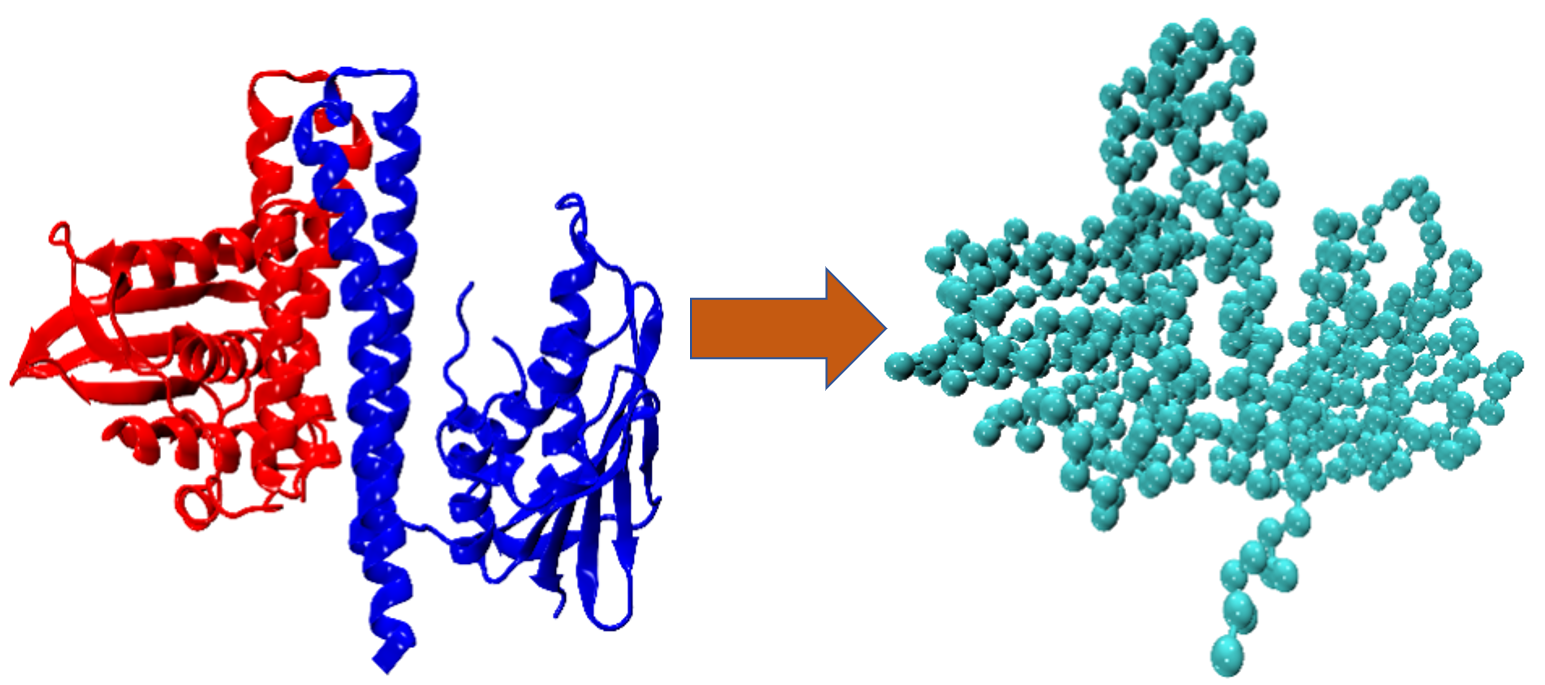
We are investigating the reversible conformational transitions between the two states using coarse-grained (CG) and atomistic molecular dynamics simulations. Our aim is to develop a framework focused on the CG level that swaps between resolution levels of description to optimize its parameterization. Specific questions include investigating different CG levels and their impact on simulation accuracy, and the impact of allowing only some of the force field parameters to be adjusted. The choice of CG method is here Go-Model. Accelerated MD integrators from project P7 will also be implemented into this framework later.
(B) QM method-DFTB: Phosphoryl Transfer Reaction step
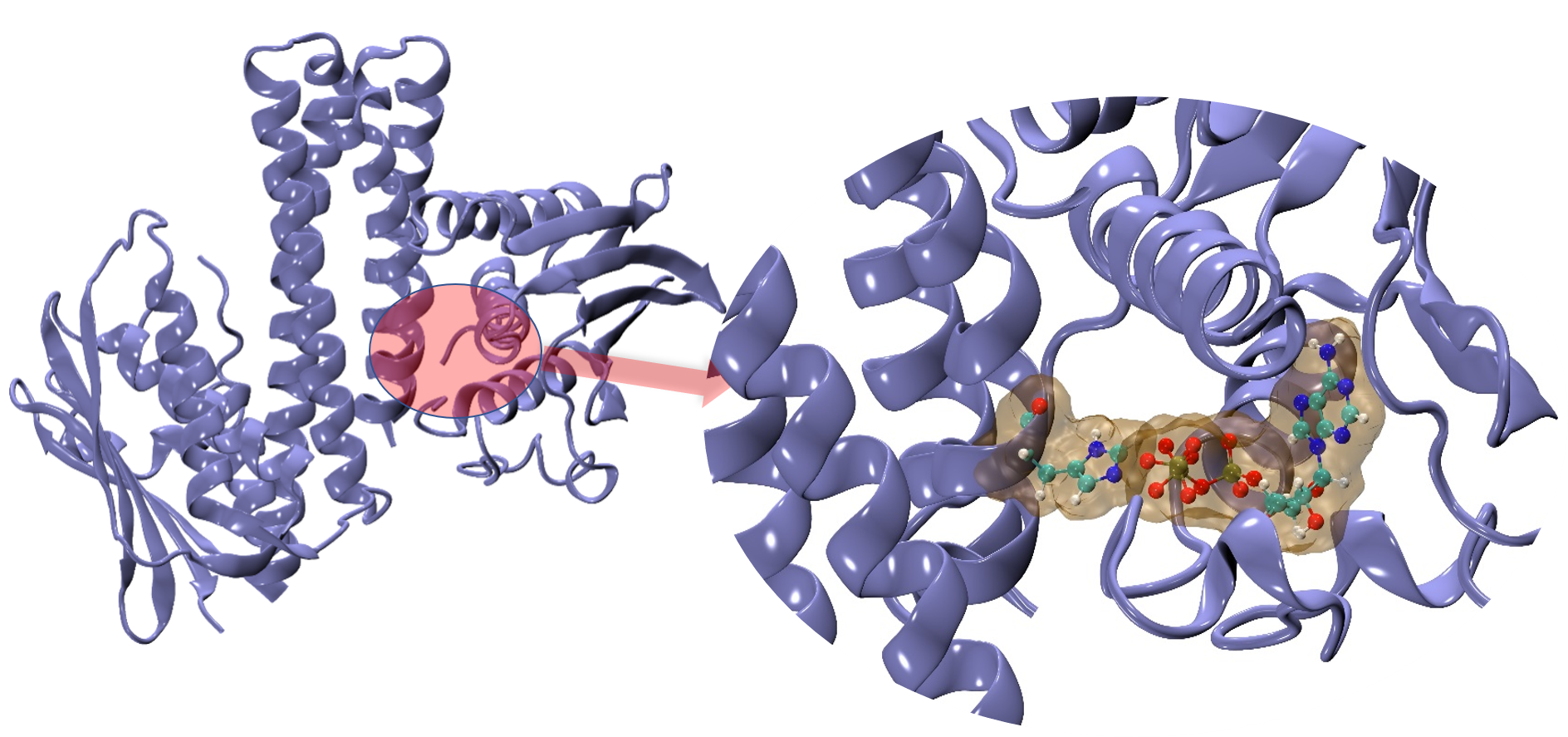
In this part we are investigating the chemical step of the enzyme. For this we need suitable QM method, our choice is DFTB (Density Functional Tight Binding Method). DFTB is a faster QM method as compared to traditional DFT or other QM methods. This allows us to make bigger QM boundaries (that means we can bigger reaction center) and we can simulate up to nano second timescale. DFTB needs accurate parameters to run, we are currently focusing on the parameterization part of it. On the other hand QM will not capture the whole picture, for example how overall protein sidechain influence the reaction, how water is playing its role in it. So, to get the big picture we need a multiscale model to study such enzymatic reaction, QM/MM which is nothing but QM and Classical Molecular Mechanics Motions are coupled with a clever equation. Once we succeed with our suitable model (all parameters are done) we will be able to describe the reaction rate and hence overall reaction mechanism.
| Name | Title | Group | Contact |
|---|---|---|---|
| Eichinger, Lena | M.Sc. | Prof. Marcus Elstner, KIT IPC (P5) | lena eichinger ∂does-not-exist.kit edu |
| 1 additional person visible within KIT only. | |||