
Dr. Denis-Mario Maag
- RTG 2450 member
- Group: Prof. Marcus Elstner, KIT IPC (P4)
- denis-mario maag ∂does-not-exist.kit edu
- www.ipc.kit.edu/tcb/english/
Multidimensional Free Energy Calculations with QM/MM Enhanced Sampling
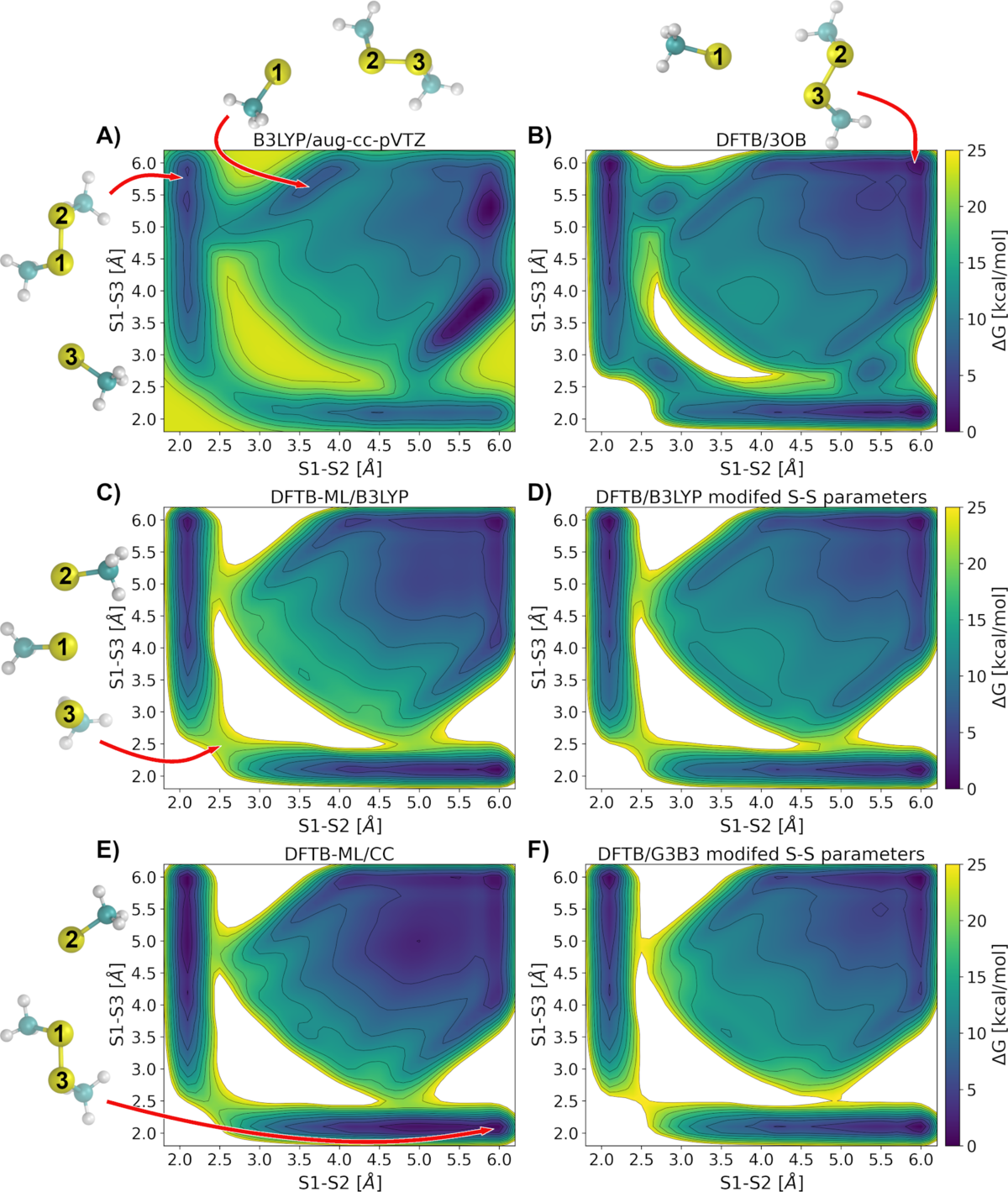
Molecular dynamics and free energy calculations of chemical reactions in which bonds are broken and formed require a quantum mechanical (QM) description. However, most QM methods are restricted to very short time scales (femtoseconds to picoseconds) due to their high computational cost. With the semi-empirical DFTB method (up to 1000x faster than DFT) it is possible to simulate such reactions on larger time scales. Just recently we used mutlidimensional QM/MM metadynamics with a simulation time of ~0.1 µs to uncover the mechanism of a long-range proton transfer in a bacterial proton pump. [Maag, D. et al. PNAS, 2021, 118 (39)]
However, the semi-empirical nature of DFTB has its drawbacks regarding certain reactions, e.g. in disulfide shuffling where artifacts at the transition states occur. These artifacts have been corrected by reparameterizing the sulfur-sulfur potential and by applying a machine learned energy correction, both with respect to higher level of theory QM methods.
Publications